1.前言
1.1水产养殖的现状及问题
我国水产养殖业是全世界发展最早的一个,同时现在也是唯一一个捕捞产量低于养殖产量的国家[1]。改革开放初期,我国水产生产链的主要方式是通过渔民或公司进行人工捕捞,一年的产量仅有121.2万t,仅占全部水产品产量的26.0%[2]。从1993年开始,我国人工养殖产量有了赶超天然捕捞产量的势头,成为我国水产品获得的主要方式,并且人工养殖在水产品总量中所占的比例仍保持较强劲的增长势头,而通过天然捕捞获得的水产品的总量依旧维持在以前的水准,也就是l400万t左右[2],水产品产量的增加主要来源于人工养殖水产品产量的增加。改革开放后期,我国提出要用“以养为主”逐渐取代“以捕为主”的方式。2011年,我国人工养殖水产品的数量达到了4023.2万t,占整个水产品的71.8%,成功完成了改革开放提出的理念[2]。石斑鱼在分类学上:目——鲈形目(鲈亚目)、科——鮨科、亚科——石斑鱼亚科、属——石斑鱼属(喙鲈属)。石斑鱼在水产养殖中也是非常重要的经济养殖鱼类,石斑鱼也逐渐成为最受渔民青睐的鱼类之一,因为其适应能力强、生长速度快、经济价值高等优点。自80年代起,网箱养殖在东南亚的印度尼西亚、文莱等国发展速度惊人。而我国是在徐绍隆等[3],在其高密度的精养研究和试验成功以后,才在我国沿海地区兴起并发展。与此同时,苗种的问题得到了有效的解决方法,更是进一步促进了石斑鱼增养殖业的发展。[5]。最近几年来,由于国民经济的飞速发展,石斑鱼的市场需求也在不断加大,养殖户为了满足市场需求和追求经济利益,在养殖方面其规模和相对养殖密度也屡创新高,这样也带来了一个严重的问题——石斑鱼疾病发生率不断上涨。而在养殖过程必须进行病害预防和治理,除了从养殖基础条件和技术(养殖环境、养殖密度)需要等到有效控制和保障外,还会被迫使用一定的药物进行疾病治理,和其他养殖业一样在石斑鱼的养殖过程中也存在抗生素滥用的情况,由此极易引起病原体产生耐受性,进一步对环境和人体健康都会构成威胁4-5]。
甚至曾有渔民不敢食用自己饲养的鱼类,就因为他十分清楚自己为了预防鱼类疾病投喂了数量甚多的药物。
由于网箱养殖石斑鱼发展迅速,衍生了一堆问题。如海水区域遭受污染、种类繁多、鱼类病害频发等都严重制约了其发展规模。其中石斑鱼病害问题尤为严重,其主要可分为三种:(1)细菌性疾病,主要分为链球菌病和弧菌病两种;(2)寄生虫类疾病,大部分仅会造成鱼类体表小创伤,死亡率低,较两外两种病害危害较小;(3)病毒类疾病[5-6],具有传染性强、死亡率高、潜伏期各不相同、症状复杂多变的特点。往往病原体都是微不可见的,直到侵入宿主体内后会以对数增长发生进行生长,此时使用抗生素类药物对其作用微乎其微;若使用化学药,往往会在未达到效果前就将鱼类本身杀死或致残。和其他物种一样能致使鱼类发病的病毒也是多种多样,不同病毒可能产生不同的症状,且病毒的传染速度和范围一般较广,而同种鱼可能成为多种病毒的感染目标,因此,病毒对鱼类的危害极其严重[7]。病毒是以寄生的方式致使鱼类生病,因病因在鱼体内,这给医治带来了巨大的困难,也为鱼类的病毒病基本无法治疗的根源,现在也还未找到有效可行的治疗方法,现在通常采取预防的方式[8]。其中对石斑鱼影响最为严重的一种病毒为神经坏死病毒,也是本实验所研究的重点。
1.2神经坏死病毒
鱼类神经坏死病毒(nervous necrosis virus,NNV),又称脑脊髓炎(Encephalonyelitis),空泡性脑-视网膜病(Vacuolating encephalopathy and retinopathy VER)属于β型诺达病毒属(Betanodavirus),同时也是目前发现的最小的鱼类病毒[9-11]。NNV的病原为病毒性神经坏死病毒,被归属于野田村病毒科中的乙型野田村病毒属,病毒粒子直径为25~30 nm,成晶格状排列在细胞质中;病毒无囊膜,呈二十面体[12-13]。病毒基因组包括两条正义的、非聚腺苷酸化的RNA单链(RNAl和RNA2)[14-15]。RNAl编码的蛋白A是病毒依赖RNA的RNA聚合酶(RdRp)的组成部分,RNA2编码病毒的衣壳结构蛋白[16-17]。Nishizawa等[17]根据衣壳蛋白的序列,将乙型野田村病毒分为了SJNNV、RGNNV、TPNNV和BFNNV4个基因型。目前神经坏死病毒的传播方式普遍认为是两种,垂直传播和水平传播[18-19]。水平传播主要是通过鱼饲养的环境、饲养所使用的器具以及投喂的饵料等一些十分基本的途径;另一种垂直传播则有两种可能的方式,一种在卵细胞中存在病毒粒子直接传给下一代,另一种则是存在于鱼的排泄物,脱落物如鳞片等的细胞中,最终可能会附着于受精卵上[20],从而传染给子代。一旦石斑鱼幼苗感染上该病毒后,死亡率达到90%以上,对于石斑鱼网箱养殖十分不利,会给渔民带来巨大的经济损失。
1.3神经坏死病毒衣壳蛋白
到目前为止,在中国所报道的NNV均属于RGNNV[21-22]。由于神经坏死病毒的衣壳蛋白在其感染宿主及其入侵细胞的时,并可诱导被感染者的免疫系统抗体的生成有着重要的作用,这意味着在外界环境重组表达神经坏死病毒衣壳蛋白来研制抗病毒疫苗具有一定的可能性[23]。陈晓艳等[21制备了一种携带NNV主衣壳蛋白的高效价(1:22000)抗血清,应该与注射途径,免疫时间间隔或次数有关。
2.实验材料与试剂
2.1实验材料
2.1.1实验菌株、载体与ULP酶
从北京全式金公司购买BL31(DE3)菌株用于大肠杆菌表达;英杰生物技术公司购得PET-SUMO质粒;ULP蛋白酶表达载体由本学院莫蓓莘老师提供。
2.1.2主要仪器与设备
本实验所需的主要仪器与设备型号及其制造商见下表2-1
表2-1主要仪器设备
仪器名称型号制造商
精密电子天平ALC-210.3 Sartorius
冷冻离心机Centrifuge 5804R Eppendorf
电热恒温水浴锅HHS-11-1上海博讯实业有限公司
恒温培养摇床THZ-300上海一恒科技有限公司
恒温培养箱LR-250上海一恒科技有限公司
超净工作台SW-CJ-1FD苏州安泰空气技术有限公司
PCR仪PTC-100 Bio-Rad
核酸电泳仪、水平电泳槽PowerPac Basic Bio-Rad
生物电泳图像分析系统FR-980上海复日科技有限公司
超纯水仪UniQUE-R20誉维公司
制冰机AF100意大利Scotsman公司
亲和层析分析仪NGC Chromatography 788-4017 Bio-Rad公司
凝胶成像系统FR-980复日科技公司
微型离心机D1008 SCILOGEX
高压灭菌锅HVE-50 HIRAYAMA公司
-80℃超低温冰箱725 ThermoFisher
吹风筒FH 020飞科有限公司
2.2实验试剂及主要试剂的配制
2.2.1实验试剂
表2-2主要试剂
名称制造商
Ex-Taq酶TaKaRa公司
琼脂糖凝胶DNA回收试剂盒TaKaRa公司
质粒提取试剂盒TaKaRa公司
IPTG上海生工
Tris-Tticine缓冲液(10x)上海生工
咪唑聚上海生工
乙二醇上海生工
卡那霉素上海生工
考马斯亮蓝上海生工
丙烯酰胺∕甲叉双丙烯酰胺(29:1)上海生工
丙烯酰胺∕甲叉双丙烯酰胺(19:1)上海生工
TEMED上海生工
过硫酸铵上海生工
透析袋北京梦怡生物
Ni离子琼脂糖GE Healthcare公司
彩色预染色蛋白标记物(10-180 KDa)GenStar公司
2.2.2主要试剂的配制
(1)100mM IPTG:称取1.1915 g IPTG白色粉末导入50ml离心管后,再以双蒸水待其完全溶解后定容,于洁净区(超净工作台)以0.22微米膜过滤除菌,放入冰箱-20℃保存;
(2)Balance Buffer:200 mM NaCl、50 mM Tris,PH调至8.0-8.1;
(3)Wash Buffer:200 mM NaCl、50 mM Tris、60 mM咪唑,PH调至8.0-8.1;
(4)Elution Buffer:200 mM NaCL、50 mM Tris、300 mM咪唑,PH调至8.0-8.1;
(5)EDTA:50 mM EDTA、20 mM NaH2PO4、50 mM NaCl,PH调至8.0-8.1;
(6)NiSO4:0.1MNiSO4;
(7)NaOH:0.5M NaOH;
(8)20%乙醇溶液:200mL无水乙醇,加去离子水定容至1L;
(9)50×TAE电泳缓冲液贮备液(pH8.0):242g Tris,51.7mL冰乙酸,100mL0.5mol/LEDTA(pH8.0),定容于1L双蒸水中;
(10)80%甘油:量取80mL的甘油,用去离子水定容至100mL;
(11)LB液体培养基:蛋白胨(5 g)、酵母膏(2.5 g)、NaCl(5g),蒸馏水0.5L,pH7.0-7.2,121℃灭20min,4℃保存。
(12)LB固体培养基:在(11)基础上再添加15%的琼脂。灭菌完毕后等待其冷却后在超净台下倒平板,注意速度要快,不然易凝固。
(13)10%APs:0.1 g(NH4)2S2O8,1ml去离子水溶解,临配临用,可存储7d。
(14)琼脂糖凝胶的制备:按照需要配制的琼脂糖凝胶体积取1..5%的琼脂粉加入到三角瓶中,并定容到相应体积。将三角瓶盖上锡纸放入微波炉中反复加热2-3次,每次一分钟并手动晃动加速溶解,直至琼脂粉完全溶解,溶液透明。
(15)聚丙烯酰胺凝胶电泳的分离、浓缩胶制备,如表2-2所示:
表2-2 SDS-PAGE胶配方
组分5%浓缩胶15%分离胶
0.5 MTris-HCl/SDS(pH8.8)–2.5 mL
1.0 M Tris-HCl/SDS(pH6.8)1 mL–
40%Acrylamide∕bisacrylamide(37.5:1)500μL 3.75 mL
去离子水2.456 mL 3.642 mL
10%APS 40μL 100 uL
TEMED(100%)4μL 8μL
Total 4 mL 10 mL
3.实验方法及过程
3.1连接PET-SUMO载体
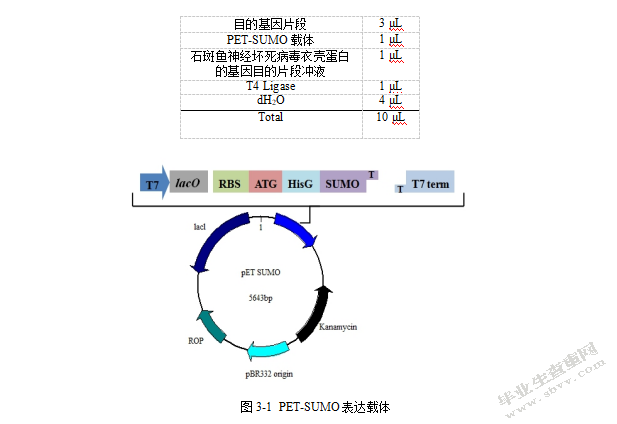
将石斑鱼神经坏死病毒衣壳蛋白的基因目标片段,以T4DNA连接酶的使用书,按步骤操作,具体如图3-1。将PCR产物插入载体,于16摄氏度水浴反应4小时,结束后4摄氏度过夜。具体连接体系见下图:
目的基因片段3μL
PET-SUMO载体1μL
石斑鱼神经坏死病毒衣壳蛋白的基因目的片段冲液1μL
T4 Ligase 1μL
dH2O 4μL
Total 10μL

3.2连接产物转化BL21菌株测序鉴定重组子
(1)在连接产物中加十倍量的感受态细胞,并轻轻混匀;
(2)①冰上放30分钟,②42度水里面放45秒钟③冰上再放5分钟
(3)新加培养基(二百微),在三十七度中培养一个小时(二百转)
(4)于加入卡那霉素的LB平板中涂布培养菌液,37摄氏度恒温培养10-16小时。单克隆菌划线,培养四到六个小时(三十七度)。
(5)质粒提取前,我先确认了Solution I中加入了RNase A,Buffer WB中加入了无水乙醇。在实验操作之前要将Solution III置于4℃预冷后使用(且实验中放在冰盒上)。我们从平板培养基上挑选单菌落接种到1-4ml的含有抗生素的液体培养基中在37℃下过夜培养。然后取1-4ml的培养菌液放入EP管中在12000rpm下离心2min,弃上清。向EP管中加入250ul溶液1用振荡器悬浮菌至无小菌块。然后,再加入250μl的溶液2并轻荡(上下)混合5-6次使菌体完全溶解直至菌液变透明。从冰盒上的Solution III中用移液枪取350ul加入EP管中,轻轻上下混合5-6次直至菌液形成紧实凝集块,然后室温下静置2分钟。将EP管放入高速离心机中以12000rpm室温下离心10分钟,取上清将核酸纯化柱置于收集管上。并将(6)中的上清液转至核酸纯化柱。将收集管及纯化柱一并起放入离心机中以12000rpm离心一分钟,弃滤液。向核酸纯化柱中加入500ul的Buffer WA,以12000rpm离心30秒,其滤液。向核酸纯化柱中加入500ul的Buffer WB,以12000rpm离心30秒,其滤液。重复步骤9。将核酸纯化柱置于新的收集管上,再放入高速离心机中以12000rpm离心1分钟,除尽残留液。将核酸纯化柱置于新的1.5离心管上,并往核酸纯化柱中央膜上加入25ul的Elution Buffer于室温下静置1分钟。将离心管连同核酸纯化柱放入高速离心机中以12000rpm离心1分钟洗脱DNA。
(6)以菌落进行上述操作,PCR扩增反应如上所述。扩增反应后,将PCR产业用1.5%AGE进行分析。具体的PCR反应体系见下表:
Ex Taq premix 12.5μL
提取pET-SUMO质粒1μL
上游引物0.5μL
下游引物0.5μL
ddH2O 10.5μL
Total 15μL
(7)测序结果表明,将反应获得的片段分子量接近相符的质粒由华大基因进行测序,从而验证目标片段在连接方向上的正确性。
3.3IPTG诱导GNN菌株诱导
(1)将在-80℃保种的菌株在冰上解冻,在超净台中按1:1000的比例即20μl菌液接种,培养过夜(三十七度,二百转/分钟)。
(2)将(1)中培养液接种(1:100),菌液:培养基(V/V)=5:500,并培养(温度=37度,转速=200);
(3)菌液培养2-3小时,OD600=0.6,冷1小时,于洁净环境中加诱导剂,条件诱导16小时(温度=18度,转速=200转/分钟)
(4)收集菌体(四度,五千转,十分钟,弃上清液),并保存(-80度),或直接进行下一的总蛋白的提取实验。
3.4诱导表达蛋白提取
(1)冰浴解冻,重悬菌体,菌体/裂解液比(W/V)1:10,裂解液配方为:200毫摩尔的氯化钠,50毫摩尔的Tris,pH=8.0-8.1
(2)总蛋白提取(超声波破碎法):超声功率25瓦,20分钟,2s(超)-4s(停)循环;
(3)总蛋白的提取,超声——离心,(1.5万转,低温,半小时),上清液为目标样品,将上清转移到新的50ml离心管中,可继续试验或保持。
3.5总蛋白SDS-PAGE胶检测
3.5.1制胶
(1)用双蒸水冲洗电泳玻璃板,晾干(吹干)后与配胶板安装后装在配脚架上,并且确保不漏液;
(2)配置分离胶:配方见表2,一定要注意的是TEMED需要在最后添加,混匀后需在短时间内灌胶,切勿过快抽打枪头产生气泡,胶装到电泳板70%的量;
(3)以1毫升水或者70%酒精轻轻的液封,一定程度上课压平胶面;
(4)大约20分钟后等分离胶凝固,在将架子向一侧切斜,将封液到去,以滤纸吸干残余封液;
(5)以表2-2的配方,配置浓缩胶,特别需要注意的是TEMED需要在最后添加,混匀后需在短时间内灌胶(用移液枪),注意不要过快抽打枪头产生气泡,需灌满;
(6)插入梳子,不能有气泡,放在那里静止不动等凝成固状,需要时间二十分钟;
3.5.2加样与电泳
(1)拿样品4倍量上样液,混匀,后开水浴,花费时间5分钟;
(2)离心时间=30s,直到管壁无液体;
(3)胶制作好的的胶装到电泳槽之中,后加入内、外液;
(4)轻取掉梳子,将已经制备好(煮好)的样品冷却,后再加入点样孔(20微升/孔),再添加蛋白标记物(4微升);
(5)电泳仪上盖,相应的正负极对好;
(6)①90伏电泳,②至四溴苯酚磺酞到交界线,③再120伏电泳,④直到溴酚蓝到凝胶下边界1到3厘米时就能停止;
(7)将胶板冲电泳仪中取下来,当心的与玻璃板剥离,用刀在二胶界限处分开,除去浓缩胶;
(8)以去离子水将玻璃板上的分离胶冲下(动作要轻),放至已经处理好的(已经过过滤)考马斯蓝染液中,并要让染液将胶淹没;
(9)将凝胶在常温下进行30分钟左右的慢速摇动,达到其充分染色的目的;
(10)拿去离子水除去凝胶上染液,脱色到染液颜色看不到还要很清楚的看到条带;
(11)用成像设备(Image Lab)成像,对可清晰明确观察到的凝胶成像并保存。脱色完的凝胶若需以后再使用可将其保存在20%的甘油中。
3.6亲和层析纯化目的蛋白
(1)将亲和层析介质装柱后,以双蒸水冲柱(填装好清河层析柱),流速=5毫升/分钟,直至cond%=0;
(2)活化柱子以5倍于柱体积(20毫升)的0.5mol的氢氧化钠,流速=5毫升/分钟;
(3)5倍于柱体积(20毫升)的双蒸水洗柱去氢氧化钠,直至cond%=0;
(4)硫酸镍过柱,至塞芬斯吸附二价镍离子到cond%稳定;
(5)双蒸水洗涤除掉未结合的二价镍离子,直至cond%=0;
(6)平衡柱子(平衡缓冲液,流速=3毫升/分钟),直到cond%稳定;
(7)样品上样;
(8)以平衡缓冲液洗去为结合蛋白,直至二百八十纳米的吸光度≈0;
(9)以添加了60毫摩尔咪唑的洗涤缓冲液洗去结合较弱的蛋白;
(10)拿洗脱缓冲液拿到目标蛋白;
(11)对目标蛋白进行透析除盐或零下80摄氏度保存。
3.7 ULP酶切
融合蛋白10毫克在加入1毫克纯化好的ULP酶(10:1),在4摄氏度下透析、酶切16小时。
3.8纯化酶切蛋白
(1)装柱后(亲和层析柱),活化柱子以5倍于柱体积(20毫升)的0.5mol的氢氧化钠,流速=5毫升/分钟;
(2)5倍于柱体积(20毫升)的双蒸水洗柱去氢氧化钠,直至cond%=0;
(3)5倍容积的硫酸镍过柱,至塞芬斯吸附二价镍离子到cond%稳定;
(4)双蒸水洗涤除掉未结合的二价镍离子,直至cond%=0;
(5)平衡柱子(平衡缓冲液,流速=3毫升/分钟),直到cond%稳定;
(6)上样,控制流速=1毫升/分钟,收集流出液,用SDS-PAGE进行分析;
(7)以5倍容积的洗涤缓冲液洗洗涤,直至二百八十纳米的吸光度稳定,用聚丙烯酰胺凝胶电泳对收集液进行分析;
(8)拿洗脱缓冲液拿到目标蛋白;
(9)对目标蛋白进行透析除盐或-80℃(加10%甘油)保存。
3.9质谱鉴定目的片段
(1)将获得的聚丙烯酰胺凝胶电泳胶上目标片段进行切胶并放在灭菌好的洁净离心管中;
(2)双蒸水(每次200-400微升)洗两遍胶,后用移液枪吸去液体;
(3)根据胶的体积加入100微升左右的考染脱色液,常温脱色1小时左右,直至胶颜色完全脱掉(无色),吸去考染脱色液)。再用双蒸水洗去剩余燃料(洗两次),以移液器吸去;
(4)脱水:50%乙腈脱色—100%乙腈将脱水,终胶呈白色;
(5)还原烷基化:添加50微升10毫摩尔的DTT溶液,溶剂2位0.5毫摩尔的碳酸氢铵,黑暗条件下反应15分钟,后用移液枪吸去液体;
(6)以10微升去离子水洗胶,后以移液器吸去液体;
(7)再次脱水,步骤与(4)一样;
(8)根据胶块大小添加5-15微升胰酶,20分钟左右胶块吸收胰酶后胀变成透明状;
(9)添加覆盖液(40微升),37摄氏度水浴16h;
(10)若需做基质辅助激光解吸电离质谱,直接取酶解的上清液上质谱;如果需要做液质联用,就必须将胶内的多肽萃取出来,具体方法为:各添加100微升的萃取液,超声10分钟,反复两次,最后冷冻干燥4小时即可;
(11)质谱鉴定结果,可通过Proteinpilot分析获得。
4.实验结果与分析
4.1石斑鱼神经坏死病毒衣壳蛋白的基因重组子测序
石斑鱼神经坏死病毒衣壳蛋白(Grouper Nervous necrosis virus capsid protein,简称gNNVcp)的基因重组子测序结果如图4-1,pET-SUMO上连接的载体序列完全吻合,这也说明pET-SUMO-gNNVcp作为目标表达载体被成功的构建出来了。

注:Sequence 0为gNNVcp基因序列,Sequence 1,2为重组子测序结果;
红色部分表示比对序列一致
4.2融合蛋白SUMO-gNNVcp的诱导表达
融合蛋白诱导表达其结果在图4-2中可见。泳道1为诱导前的总蛋白条带,泳道2为诱导后的条带。由图可看出诱导后的总蛋白条带在接近50kDa处较诱导前的条带有明显增粗,说明诱导后有显著增多的目的条带(SUMO-gNNVcp理论值为50kDa),因此可初步确定本实验成功表达出石斑鱼神经坏死病毒衣壳蛋白融合蛋白。
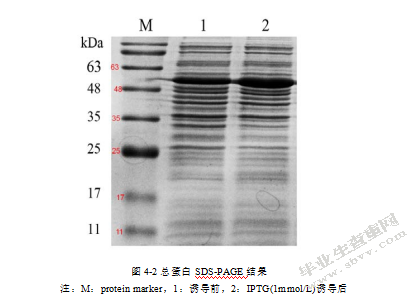
图4-2总蛋白SDS-PAGE结果
注:M:protein marker,1:诱导前,2:IPTG(1mmol/L)诱导后
4.3融合蛋白酶切纯化
将上述SUMO-gNNVcp融合总蛋白经亲和层析纯化后进行ULP酶切过夜,透析浓缩,再经过亲和层析纯化后SDS-PAGE得到如图4-3所示结果,泳道1为融合蛋白酶切后,泳道2为融合蛋白酶切后纯化后,其对应条带的位置与预期相同,gNNVcp目的蛋白约为37KD,图中35kDa与48kDa之间的条带为酶切后释放出的目的条带,小于17kDa的那一条带为伴侣分子SUMO蛋白。可见SUMO-gNNVcp融合蛋白已被ULP酶切开且经亲和层析后显示为单一目的条带,因此本研究获得高纯度的抗原蛋白gNNVcp。
4.4目的蛋白gNNVcp的质谱鉴定
将亲和层析纯化后通过切胶回收获得的的gNNVcp目的蛋白送深圳大学质谱室鉴定得到以下结果,绿色氨基酸表示检测到与斜带石斑鱼gNNVcp(Epinephelus coioides nervousnecrosis virus coat protein,GenBank:AAN04042)序列完全一致的片段,覆盖率高达97%,表明gNNVcp目的蛋白成功表达。质谱峰图见后。
MVRKGEKKLAKPATTKAANPQPRRRANNRRRSNRTDAPVSKASTVTGFGRGTNDVHLSGMSRISQAVLPAGTGTDGYVVVDATIVPDLLPRLGHAARIFQRYAVETLEFEIQPMCPANTGGGYVAGFLPDPTDNDHTFDALQATRGAVVAKWWESRTVRPQYTRTLLWTSSGKEQRLTSPGRLILLCVGNNTDVVNVSVLCRWSVRLSVPSLETPEETTAPIMTQGSLYNDSLSTNDFKSILLGSTPLDIAPDGAVFQLDRPLSIDYSLGTGDVDRAVYWHLKKFAGNAGTPAGWFRWGIWDNFNKTFTDGVAYYSDEQPRQILLPVGTVCTRVDSEN
5.讨论
1.本研究选择以缺失A的粘性末端的pET-SUMO载体为原核表达载体,因其能直接和经Taq预混酶PCR扩增获得的片段相连,可相对容易构建获得工程质粒。
2.pET-SUMO载体上构建有经SUMO修饰蛋白,这可大幅提高表达蛋白融合性,从而使类似于OmpK如此的大分子蛋白也可以在原核中成功表达;
3.为避免宿主排斥外来基因的表达,在pET-SUMO载体插入了,更有利于表达载体表达的噬菌体T7操纵子。
4.根据Sumo载体的特性:(1)Sumo表达载体具有His标签,可用亲和层析分离纯化融合蛋白。(2)这个表达载体的特殊性还在于配备了ULP酶,该酶能特异性识别sumo泛素蛋白与目的蛋白之间的两个甘氨酸位点,进而切除His标签以及sumo泛素蛋白,通过再一次亲和层析纯化出纯度极高的目的蛋白。这可进一步降低杂蛋白的影响,为后续试验及以后的大规模生产,工业化等提供了极大的便利。因此,在表达试验中,想获得更多的目标蛋白,则需要在试验过程中做到极其细化,从而获得最佳条件。可以通过以下几种方法:1、在前人基础上尝试更多的OD值下的诱导试验,2、细化诱导时间(如时间间隔设为1小时或半小时)表达量的观察,3、加大表达温度和转速的梯度。
5.在后期进行对幼苗注射实验时可能需要从多方面进行实验,来确认该产品的实用性以及最佳生效条件。
6.结论
本实验将合成的神经坏死病毒目的基因与载体结合,通过转入高效表达菌BL21,表达出相应蛋白,得出所需目的条带,最后使用质谱方法验证蛋白表达正确,成功制备相关神经坏死病毒抗原。通过上述方法,在实验中得出成功构建pET-SUMO-gNNVcp载体和该载体在大肠杆菌中原核表达成功结论,揭示了所示方法对gNNV蛋白产品的规模化生产的可行性,为制备具有生物有效性抗体打下了坚实的基础。
【参考文献】
[1]纪春艳.浅析我国水产养殖业的发展现状[J].渔业致富指南,2016(22):14-16.
[2]刘佩,孙炜琳.我国水产养殖业的发展现状·存在问题及对策[J].安徽农业科学,2013(30):11981-11984.
[3]林超辉.斜带石斑鱼的生物学特性及养殖技术[J].水产科技情报,2007,34(5):206-207,209
[4]华鼎可,张永嘉,黎祖福,等.石斑鱼膨胀病(打转病)、溃疡病、烂尾病的防治研究[J].鱼类病害研究,1990,12(4):26-28.
[5]罗鸣,陈傅晓,刘龙龙,等.我国石斑鱼养殖疾病的研究进展[J].水产科学,2013,32(9):549-554.
[6]杨亚惟,冯乾生.石斑鱼的常见疾病及诊治[J].现代畜牧科技,2017(1):51-51.
[7]张奇亚.我国水生动物病毒病研究概况[J].水生生物学报,2002,26(1):89-101.
[8]黄琪琰.水产动物疾病学[M].上海:上海科学技术出版社,1993:65.
[9]黄剑南,林蠡,翁少萍,等.赤点石斑神经坏死病毒外壳蛋白全基因克隆与序列分析[J].水产学报,2005,29(3):429—432.
[10]GROVE S,JOHANSEN R,DANNEIG B H.et a1.Experimentalinfection of Atlantic halibut Hippoglossus hippoglossus with nodavirug:tissuedistribution and immune response[J].Dis Aquat Ors,2003,53(3):211—221.
[11]Chi S C,Lo B J,Lin S C.Characterization of grouper nervous necrosis virus(GNNV)[J].Journal of Fish Diseases,2001,24(1):3–13.
[12]Comps M,Pépin J F,Bonami J R.Purification and characterization of two fish encephalitis viruses(FEV)infecting Lates calcarifer,and Dicentrarchus labrax[J].Aquaculture,1994,123(1–2):1-10.
[13]Mori K,Nakai T,Muroga K,et al.Properties of a new virus belonging to nodaviridae found in larval striped jack(Pseudocaranx dentex)with nervous necrosis.[J].Virology,1992,187(1):368-71.
[14]Ball L A,Johnson K L.Reverse genetics of nodavimses[J].Advances in Virus Research,1999,53:229—244.
[15]Schneemann A,Reddy V,Johnson J E.The structure and function of nodavirus particles:a paradigm for understanding chemical biology[J].Advances in Virus Research,1998,50(1):381-432.
[16]Nagai T,Nishizawa T.Sequence of the non-structural protein gene encoded by RNA1 of striped jack nervous necrosis virus.[J].Journal of General Virology,1999,80(Pt 11)(11):3019.
[17]Nishizawa T,Mori K,Furuhashi M,et al.Comparison of the coat protein genes of five fish nodaviruses,the causative agents of viral nervous necrosis in marine fish.[J].J.gen.virol,1995,63(7):1563-1569.
[18]Iwamoto T,Mise K,Mori K,et al.Establishment of an infectious RNA transcription system for Striped jack nervous necrosis virus,the type species of the betanodaviruses.[J].Journal of General Virology,2001,82(Pt 11):2653-2662.
[19]Grove S,Johansen R,Dannevig B H,et al.Experimental infection of Atlantic halibut Hippoglossus hippoglossus with nodavirus:tissue distribution and immune response[J].Diseases of Aquatic Organisms,2003,53(3):211.
[20]林克冰,方琼珊,吴建绍,等.石斑鱼神经坏死病毒传播途径阻断的初步研究[J].渔业研究,2011(5):15-19.
[21]陈晓艳,何建国.斜带石斑鱼神经坏死病毒主衣壳蛋白抗体的制备[J].中国水产科学,2006,13(5):841-844.
[22]田飞焱,吕建强,刘荭,等.6株鱼类病毒性神经坏死病病毒cp基因的分子特征和遗传发生分析[J].华中农业大学学报,2008,27(3):414-418.
[23]Hegde A,Chen C L,Qin Q W,et al.Characterization,pathogenicity and neutralization studies of a nervous necrosis virus isolated from grouper,Epinephelus tauvina,in Singapore.[J].Aquaculture,2002,213(1–4):55-72.
【Abstract】The aquaculture industry currently plays a decisive role in our country and is closely related to our lives.However,there have been many problems in the development of aquaculture.Nerve necrosis is a virus disease that is harmful to cage cultured grouper,and the survival rate of seedlings is very low once it is affected.Therefore,research on the neuronecrosis virus has become a very hot topic,and it is also an important application direction of genetic engineering and protein engineering in the aquaculture industry.This study is an extension of the work under this topic.In this experiment,the target gene of neural necrosis virus in grouper was transplanted into E.coli by fusion expression,and E.coli was able to express the virus synthesis protein by SDS-PAGE and Western-blot.The specific operation is as follows:
(1)In this study,the recombinant expression plasmid vector pET-SUMO containing(Grouper Nervous necrosis virus capsid protein,abbreviated gNNVcp)gene was first transformed into E.coli BL21(DE3)for fusion expression;
(2)After fusion expression,we identified the protein synthesized by E.coli by SDS-PAGE and Western-blot,and confirmed that the grouper nerve necrosis virus capsid protein was expressed in recombinant E.coli.After this point,we first purified the protein by affinity chromatography.
(3)In this experiment,the protein was subjected to ULP digestion and affinity chromatography after purification by affinity chromatography,and the fusion expression protein with a degree of purification as high as 97%was obtained through mass spectrometry analysis.Therefore,this experiment confirmed that E.coli can express the grouper nerve necrosis virus capsid protein in groupers.At the same time,the purity of the viral protein can reach 97%after further purification.Therefore,it has been concluded that grouper can be made by this method.The conjecture of a vaccine for neuronecrosis virus.
【Key words】grouper;nervous necrosis virus;Expression vector of pET-SUMO;preparation of antigen;recombinant expression
下载提示:
1、如文档侵犯商业秘密、侵犯著作权、侵犯人身权等,请点击“文章版权申述”(推荐),也可以打举报电话:18735597641(电话支持时间:9:00-18:30)。
2、网站文档一经付费(服务费),不意味着购买了该文档的版权,仅供个人/单位学习、研究之用,不得用于商业用途,未经授权,严禁复制、发行、汇编、翻译或者网络传播等,侵权必究。
3、本站所有内容均由合作方或网友投稿,本站不对文档的完整性、权威性及其观点立场正确性做任何保证或承诺!文档内容仅供研究参考,付费前请自行鉴别。如您付费,意味着您自己接受本站规则且自行承担风险,本站不退款、不进行额外附加服务。
原创文章,作者:写文章小能手,如若转载,请注明出处:https://www.447766.cn/chachong/14375.html,