1文献综述
1.1氨噻肟酸
1.1.1目前,我国氨噻肟酸生产技术逐步成熟,产量随之增加,但就成品质量及技术工艺而言,其存在的问题不容小觑。当下鲜有研究人员对于氨噻肟酸脱水重结晶及反应结晶两个具体步骤进行研究,但成品的收率及质量以结晶技术的优劣为前提。本研究以提高工艺技术为目的,系统地对氨噻肟酸脱水重结晶及反应结晶两个过程进行分析和探讨,旨在实现既使成本减少,又保障成品的收率和质量的双赢局面。20世纪80年代中期,研究人员对氨噻肟酸类化合物进行研究时惊喜地发现,由其合成的头孢菌素的抗菌性十分可观,且其毒性和副作用相对较小。由此,氨噻肟酸类化合物被视为抗生素侧链而广泛地用于临床工作,该物质也因此成为重要的医药中间体[2,3]。不少相关公司有计划或正在安装生产氨噻肟酸的相关设备以增加头孢他美(酯)、头孢地嗪、头孢噻肟及头孢三嗪等头孢类抗生素的产量。这一巨大市场极大地提高了氨噻肟酸在国内的价值和地位。氨噻肟酸在2000年时的国内产量为400吨左右[4],其主要用途是生产头孢菌素类半合成抗生素所需的AE-活性酯,其中一部分作为潜在价值颇高的医药中间体出口国外[5]。但国内现有的生产工艺存在反应步骤多、周期长、三废多喝收率低等缺点。改进的生产工艺大大缩短了工艺步骤,简化了操作,缩短了反应周期,降低了生产成本,具有很好的工业价值。
1.1.2产品介绍
氨噻肟酸,化学名称2-(2-氨基-1-噻唑基)-1-2-甲氧亚胺乙酸(顺式)。
英文名称:(2)-2-methoxyimino-2-(2-aminothiazole)-4-acetic acid。综合文献[6]。英文缩写:ATMAA;ACX Number X1032998-3,分子式C6H7N3O3S,分子量201.2。CB Number:CB5294497。
熔点:134-150℃;水溶解性:0.6g/100ml(20摄氏度);CAS数据库:65872-41-5(CAS Data Base Reference);危险品标志:x1;危险类别码:36/37/38;安全说明:26-37/39-24/25。
性状:产品分为含结晶水和不含结晶水两种。含结晶水产品为白色或淡黄色结晶粉末,产品在多数有机溶剂中不溶,可溶于热水、强碱、强酸溶液中,高温分解并放出硫化氢气体。基础物质含量≧87%,含水量≦13%。其上游原料为硫脲,下游产品为头孢噻肟钠。
1.1.3氨噻肟酸的用途
氨噻肟酸既是多种头孢类抗生素的中间体,如头孢三嗪(头孢曲松)、头孢双唑、头孢噻肟钠等;又是诸如RH756头孢氨噻三嗪等物质的侧链。氨噻肟酸可以进一步加工成AE活性酯中间体[7]。特别是头孢噻肟钠和头孢三嗪等以列入国家基本药物,头孢噻肟还是基本医药保险用药。
1.2氨噻肟酸发展历史
1.2.1头孢类药物及中间体的发展
氨噻肟酸是能够合成AE-活性酯的一种医药中间体,它在第三代头孢菌素的合成过程中功不可没。
1985年,两名研究人员对4-氯乙酰乙酸乙酯进行肟化、环合、甲基化三个过程后,生成了氨噻肟酸乙酯,然后对这一中间产物进行皂化水解后最终得到氨噻肟酸[8]。
弗莱明是第一位在抗生素中发现青霉素的英国学者,于1929年发现。随后,某英国高校的专家对青霉素实施了纯化和分离的工作,同时发现它在传染病治疗中颇为有效。
随后抗生素的生产和应用得到了快速发展。第一代头孢类抗生素-头孢噻吩,由XE.Lily公司生产并于1964年上市。截至目前,第四代头孢类抗生素已经问世。依据问世时间的早晚与不同药理作用而划分的每一代抗生素分别具有各自不同的功能。
据统计,2007年全球抗生素占有很大的份额。2008年,全球有65种头孢类抗生素药品上市。作为更新换代最快的抗生素,它在国内市场的占比已高达13%以上。在头孢泊肟酸、头孢匹罗、头孢噻呋钠等药物组成成分中可发现氨噻肟酸的身影[9]。在医药事业蓬勃发展的今天,医药中间体的产量也随之大幅增加。在国内,用于医药生产的中间体与化工产品可大致实现自行配套,我国可生产360多种医药中间体,其中用于出口的为220种左右,平均年产量高达3万t左右。此外,仍有巨额支出用于医药中间体的进口工作。
日前,头孢类抗生素在国内拥有前所未有的发展前景,各大型制药企业诸如石家庄制药集团、珠海丽珠集团、哈尔滨制药总厂、华北制药集团、珠海联邦集团等均大幅增加其生产数量。在这一巨大浪潮的涌动之下,药物中间体的需求空前增大,这意味着其产量亦要随之大幅增加。因此,我国在诸多生产国之中独占鳌头,成为世界最大的中间体生产国家。
当下我国生产最多的四种抗生素侧链中间体分别为氨噻肟酸、D-对羟基苯甘氨酸及其邓钾盐、三嗪环。各个氨噻肟酸的生产商在生产技术逐步成熟的基础上逐渐扩大生产规模和数量,且源于各行各业的资本也不断注入到这一产业中,为其提供了更大的活力。由于我国更好地平衡了减少成本与增加产量之间的关系,不少国外医药的采购量较前明显增加。国内氨噻肟酸在2003年的产量为1200t,六年后则超过了3000t,实现了巨大进步。石家庄合佳保健品有限公司、河北金通医药化工有限公司、山东金城医药化工股份有限公司、浙江普洛得到邦制药有限公司等是氨噻肟酸的主要生产厂家。国内的氨噻肟酸以合成头孢菌素为主要目的,在少量出口的基础上呈增加趋势。
如2000年出口量为48吨,2001年出口量为70吨,2002年超过120吨,2003年达170吨。由此可见,以氨噻肟酸为侧链的头孢菌素在治疗疾病方面表现优异,因此其出口量随之快速增加。换言之,在生产技术逐步成熟、成本日渐缩小的基础上,市场竞争亦愈演愈烈。日前,不少研究人员依然对以氨噻肟酸为中间体的新型抗菌素进行研究,因此其需求量依然不减。
1.2.2我国氨噻肟酸及其下游产品的开发和生产情况
随着当前国内头孢类抗生素的前景日渐繁荣,氨噻肟酸也因此得到巨大发展。自二十世纪末期开始,许多诸如四川天然气研究所、国家医药管理局上海医药工业研究所、河北轻化工学院、天津大学及浙江大学等研究机构纷纷对其进行深入的研究和分析,并在前人的基础上对不完善的地方予以进一步地修改和优化。经过近20年的实践,生产技术已经基本定型,目前国内主要采用乙酰乙酸乙酯为原料生产。由于氨噻肟酸在被合成环合物前保持的液体状态难以严格计算,虽然薄层层析的方法已然得以应用,但对其进行质量分析以及中间状态的控制还不尽人意,因此还尚未能够对生产过程起到指导性意义。现代科技不断发展,高压液相分析技术逐渐成熟完善,利用率也越来越高,因此以其对氨噻肟酸的生产过程采取监控措施已经成为可能,在此基础上,生产投料技术则得到了进一步的改良和指导。当下我国生产氨噻肟酸的投料比是确定的,若计算不当,则会造成浪费原料的严重后果。因此,处理好生产过程中例如计算比例等相关检测问题对于其提高产量、节约成本显得很重要。
氨噻肟酸生产成本高的关键因素是溴化反应的高危险性,且溴是在整个生产过程中的中间产物,在成品当中并不存在。所以,采用危险小、成本低的原料(如液氯)则会对生产过程起到极大的帮助。尽管以液氯替代溴的使用已被某德国公司创下先例且为其申请了相关专利,但对于这一使用方法的相关研究和报道仍然十分罕见。溴化,是由单一溴原子参与反应的反应过程,另一个则以溴化氢的形式以便于进一步生成氢溴酸或溴化钠而被回收。关于单质溴可由溴离子氧化而成,随后重新参加化学反应的相关研究已经面世。
反应环境不同,氨噻肟酸的结构也随之变化(其中反式结构并无治疗效果)。所以在制备过程中应万分留意其两种结构比例,以最大地提高其有效率。精制过的氨噻肟酸鲜有杂质。但因其羧基、氨基并存的特殊结构,使得其内部发生缩合反应的概率增大,从而生成对制药不怎么友好的二聚体结构。
目前,出口产品往往要求检测顺反异构体和二聚体的含量。在AE-活性酯的生产中利用价格较低的原料代替昂贵的三笨基磷新工艺已在韩国实现了工业化生产,国内现在各生产厂家均在密锣紧鼓的研制该工艺(目前已经有几家企业进行了试生产),AE-活性酯的标价将会随着这一工艺的逐步成熟而日渐下降,如此则更进一步地促进了头孢他美酯、头孢三嗪等头孢类抗生素在国内的大量生产。
河北科技大学在上世纪末对氨噻肟酸合成技艺进行改良,以二氯甲烷(原为乙酸乙酯)参加亚硝化后的萃取反应液这一化学反应,在室温4小时的条件下,相关转移催化剂为前提,进行甲基化反应并定量分析了肟化产物,最终在诸多实验结果中确定了1:1.2是硫酸二甲酯和肟化物的最优比。以相关转移催化剂在两相介质中进行甲基化反应的优良工艺,不仅保障了这一反应的充分性、便捷性,又降低了相关反应原料的使用量,节约了成本。一条年产量为30吨左右的氨噻肟酸生产线在1997.03由河北某医药化工企业建立,这一生产线使用的正是此工艺技术。在随后的三年时间里,氨噻肟酸的生产装置增至100t/a。s在接下来的一年时间里,去甲氨噻肟酸、2,4-氨唑二酮、头孢他啶活性酯、氨基噻唑乙酸、头孢他啶侧链酸、等系列产品逐一问世,AE-活性酯的生产设备也增至与氨噻肟酸的同等数量。国内氨噻肟酸生产技术、产量在头孢菌素这一巨大浪潮的推动之下得到了前所未有的发展,多家公司争相安置其生产设备。21世纪初,国内主要的氨噻肟酸生产商有临海新星化工厂、四川天然气研究所、浙江永宁制药厂、辽宁抚顺美强化工厂、河北金通医药化工有限公司、浙江横店永安-得邦有限公司、以及石家庄诚业化工厂等等,总产量为400吨左右。这些厂家生产的是以合成生产头孢菌素的AE-活性酯为主要目的的氨噻肟酸,其中小部分用于出口。随着肥城市鑫源化学原料有限公司氨噻肟酸生产装置的更新升级,我国的生产力已高达1000t/a。
尽管如此,我国对于氨噻肟酸下游产品的研究技术尚不成熟,与国外相差甚远,详见表1.1。
表1.1国内外头孢菌素的技术指标差距(氨噻肟酸下游产品)
品种国内收率(%)国外收率(%)
头孢噻肟一般75 80
头孢曲松一般50 60
头孢他类-60以上
国内对于氨噻肟酸合成的下游产的品种生产较少,“十五”开发规划所包含的主要有以下几种,详见表1.2。
表1.2“十五”推荐研究开发的氨噻肟酸下游产品
名称简介
头孢他美酯第三代口服头孢菌类。被国家级化学医药新产品指南收录。
头孢地尼口服头孢菌类新药,相对于头孢克肟而言,了对G菌(包括MRSA在内)的活性有所提升。国内依据进口使用,并不生产此药,被国家级化学医药新产品指南收录
头孢唑南第四代注射头孢菌类,2005年专利保护期满。是“九五”计划研制的品种,需要继续研制。
头孢克肟第三代口服头孢菌类。被国家级化学医药新产品指南收录。
头孢托仑酯口服头孢菌类新类型。国内依据进口使用,对G菌的疗效有所提升
头孢匹罗第四代口服头孢菌类。该品种被列入“九五”研制计划,需要继续研究。
1.3氨噻肟酸合成工艺
目前现存的氨噻肟酸合成方法主要有四种,分别是:4-氯乙酰乙酸酯法、乙酰乙酸乙酯单步法、乙酰乙酸乙酯一锅煮法以及乙酰乙酸甲酯法。其中我国使用的主要是第二种方法。但这一方法存在诸多不足,如反映周期长、中间产物量无法确定、反应收率低、成本体积大、有机溶剂用量大等等。原有的生产技术存在颇多不足,如无法对反应进程随时监测,不利于生产的二聚体依然存在,生产成本依旧较高等等。所以使技术更优良、有效率更高是当下亟待解决的问题。
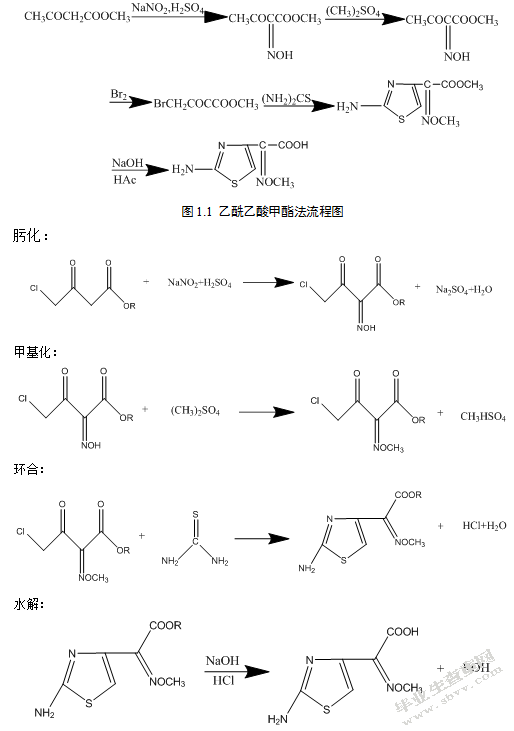
1.3.1乙酰乙酸甲酯法
由乙酰乙酸甲酯与亚硝酸钠发生肟化反应制得2-羟肟乙酰乙酸甲酯,与硫酸二甲酯反应制得2-甲氧亚胺乙酰乙酸甲酯,以上这些化学物质通过一系列的化学反应,最终得到乙酸乙酯,该物质又可以与硫酸反应得到其他的物质,例如甲氧亚胺乙酰乙酸和甲氧亚胺乙酸甲酯,所得产物用氢氧化钠进行皂化,再用盐酸中和得到氨噻肟酸,分为肟化、甲基化、溴化、环合、水解五步,流程如下:

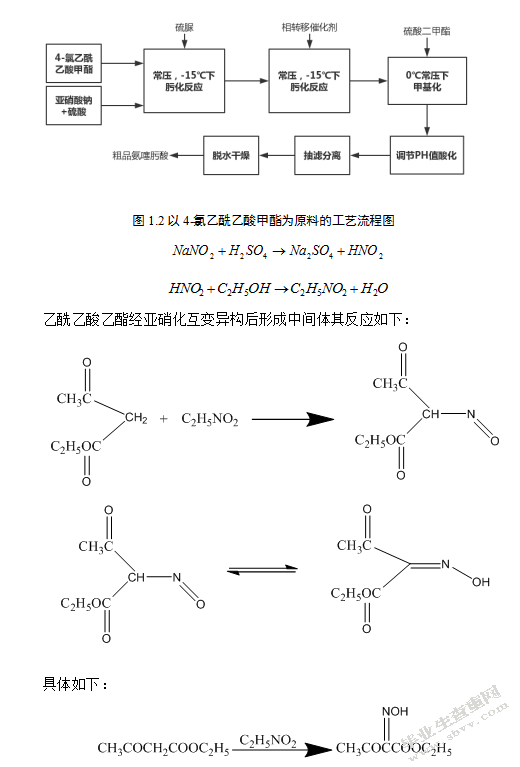
1.3.2 4-氯乙酰乙酸酯法
氯乙酰乙酸酯法有很多优点,它的方法是可直接用4-率乙酰乙酸甲酯和亚硝酸钠肟化得到4-氯-2-羟肟乙酰甲酯发生化学反应得到乙酸甲酯,由于乙酸甲酯容易被氧化,所以在常温条件下下,它能硫脲反应得到甲氧亚胺乙酰甲酯,在一定条件下,比如将它皂化之后,用盐酸进行中和反应,就会在短时间内产生氨噻肟酸,反应式如下:
以4-率乙酰乙酸甲酯为原料的工艺流程图如图1.2所示:
1.3.3乙酰乙酸乙酯一锅煮法
常见的制备氨噻肟酸的方法是蒸馏制备法,既将氨噻肟酸单独放在一个封闭的锅内,让蒸馏水和它进行水解,风化以及环合,这种方法制备氨噻肟酸收益率非常高。
此法若能成功应用于工业生产,则会大幅度的降低生产成本,但此法尚未成熟。
1.3.4乙酰乙酸乙酯单步法
我国现在以相对完善的乙酰乙酸乙酯单步法为制造氨噻肟酸的主要技术。这一技术将乙酰乙酸乙酯经过五个步骤,最终生成氨噻肟酸。这五步分别为肟化-甲基化-卤化-环合-水解[10-16]。
(1)肟化反应
以乙酰乙酸乙酯、亚硝酸乙酯气体为原料在5-8℃常压条件下搅拌反应,制得中间体2-羟肟乙酰乙酸乙酯,亚硝酸乙酯气体由亚硝酸钠和稀硫酸、乙醇制备,其反应方程式如下:
乙酰乙酸乙酯经亚硝化互变异构后形成中间体其反应如下:
具体如下:
亚硝酸乙酯(以气体存在)对反应条件要求较高,其温度需适中(室温适宜),过高则后果不堪设想,过低则反应速率不足而致反应不充分,剩余的原料经后续化学反应会产生新的产物,方程如下:
(2)甲基化反应
将适量的硫酸二甲酯加入到上步所得的2-羟肟乙酰乙酸乙酯中,搅拌混匀使两者充分接触,混匀后分别加入乙酸乙酯、无水硫酸钠以进行抽提、干燥,中间经酯层水洗,最后蒸去溶剂,分别加入氯仿、正己甲烷重结晶,得2-甲氧亚胺乙酰乙酸乙酯。具体如下:
(3)溴(氯)化反应
用溴素或硫酰氯将2-甲氧亚胺乙酰乙酸乙酯溴化或氯化得到4-溴(氯)-2-甲氧亚胺乙酰乙酸乙酯,在反应过程中会产生较多溴化氢,这种气体会在很大程度上减慢反应进度和效率,使得反应不完全,因此反应过程中应及时抽掉反应气体,不仅如此,为保证后续环合反应的顺利进行,在溴化或氯化反应完成后,为确保溶液中溴化氢气体被最大限度地去除,应向溶液中通入空气。
(4)环合反应
硫脲与(3)得到的4-溴(氯)-2-甲氧亚胺乙酰乙酸乙酯粗品经缩合反应生成取代异硫脲中间体,随后分别经过脱水、环合反应最终生成一对顺反异构体。具体如下:
图1.3生成一对顺反异构体的反应式
待反应完全,加入Na2CO3以控制PH范围,沉淀滤出后,分别经过抽滤、洗涤、干燥三个过程,最终得到2-(2-氨基-4-噻唑基)-2-甲氧亚胺乙酸乙酯。
(5)水解反应
使用NaOH皂化2-(2-氨基-4-噻唑)-2-甲氧亚胺乙酸乙酯,随后以醋酸中和,得到成品氨噻肟酸,具体如下:
图1.4氨噻肟酸生产工艺流程图
虽然我国使用的乙酰乙酸乙酯单步法已经相对成熟和完善(其制作过程已在上图1中展示),但仍然存在对环境不友好、有机溶剂需求高、后期处理过于繁琐以及危险性大等弊端。作为一种强放热反应,卤化反应要求以足够完善的物料配比和冷却系统为必备的基础设施,不然则会发生后果不堪设想的爆炸事故,损失不可估量。因此,这一反应是生产过程中危险性最高的步骤。此外,溴素作为原料之一,具有高成本、高毒性的两大特点,且同时具有氧化性,因此选择其他产品替代这一原料以节约成本、提高技术、降低毒性成为各大厂商的不约而同的做法。本研究以乙酰乙酸乙酯单步法为研究方法。
2实验部分
2.1实验步骤
(1)肟化、甲基化反应
将0.6mol(78g)的乙酰乙酸乙酯、0.65mol(45g)亚硝酸钠及200毫升水按顺序加入到反应容器中。在搅拌的同时将24毫升浓度为98%的硫酸与100毫升水的混合溶液滴加进去,在保证温度在0~5℃之间的基础上搅拌120分钟。
将24mL98%的硫酸加入到100ml水中,混匀放置一旁备用,先后分别将78g乙酰乙酸乙酯、45g亚硝酸、200mL水加入反应瓶中,缓慢搅拌,将上述硫酸溶液逐滴加入其中,滴完后继续搅拌2h,整个过程在2.5±2.5℃温度下进行,搅拌完成后,分3次用200mL二氯甲烷萃取。有机相似饱和食盐水洗涤,再将1g四丁基溴化铵、76gNa2CO3、150mL水加入萃取液,其后将环境温度设置在30℃,滴入90.7g(CH3)2SO4,反应2h。将溶液静置片刻,待其将水相分去。在用600ml有机相似水分洗涤两次后,用无水Na2SO4将其干燥后静置一晚。得到98.5g的粗2-甲氧亚胺乙酰乙酸乙酯成品,在低温条件下保存以备后续使用。收率95%。
(2)溴(氯)化
取(1)的产品69.2g(0.4mol)溶于40mLCH2Cl2,放入反应瓶中,加入11mL N,N-二甲基甲酰胺。在室温条件下,将64.8gSO2Cl2逐滴加入进去。随后待温度升至40℃后继续搅拌混匀,让其充分反应600分钟。反应毕,缓缓加入60mL水,静置,自然冷却分层。水相似10mLCH2Cl2抽提,合并有机相,以无水硫酸钠干燥。减压浓缩除去溶剂,得淡黄绿色油状液体76.5g。收率92.2%。
(3)环合
将60ml乙醇与62.3g第(2)步所得的黄绿色油状液体充分混合以待备用。分别将120mL水、27.4g硫脲与60mL乙醇分别加入容器中混合30min,待其温度下降至15℃(及以下)后滴入上述的混合溶液。
加完后,继续搅拌反应3h。整个过程控制温度低于15℃。反应毕,以氨水调PH值至7,继续搅拌反应4min,抽滤。用干净水源将滤饼洗净。在真空且温度控制在50℃以下的干燥环境中得到46.1g类白色粉末状结晶。收率60.8%。
(4)水解
将50ml乙醇与9gNaOH混合,然后取45.8g(3)步所得的粉末状结晶加入进去,在温度控制为40~45℃的条件下混合搅拌60分钟。待溶液变透明,把容器放进冰浴中冷却,冷却至温度约0℃时加入冰乙酸将其pH调至6直到完全结晶。将其快速抽滤,并将母液回收以备后续使用。将乙醇与水按照1:1的比例混匀,用该溶液清洗滤饼,随后用乙醚进行二次清洗后待其冷却,按照上述方法进行处理后可得7.0g淡黄色粉末状结晶。氨噻肟酸产品累计获得35.3g。收率84.5%,总收率45%。
2.2HPLC仪器
2.2.1HPLC概念
高效液相色谱法(HPLC),别名有“高分离度液相色谱”、“高效液相色谱”、“近代柱色谱”“高速液相色谱”等。它的分析方法如下:液体为流动相,通过高压输液系统把缓冲液、单一溶剂或混合溶剂等液体加入到色谱柱(已装固定相)中进行进一步分离,随后对其进行检测以得到分析结果。工业、法检、化学等多个学科领域已将这一技术作为分析分离的主要方法之一,得到广泛应用。
2.2.2HPLC特点
高效液相色谱法有一下特点:
(1)高压:以液体为流动相,需采取高压措施以抵抗其经由色谱柱时受到的阻力,从而达到加速通过的效果。
(2)高速:相比于传统的分析方法而言,HPLC的载液流速及分析速度有了更大的提升。例如某些样品的分析速度可快达5分钟,一般样品分析时间为一刻钟到半小时,大多数在60min内完成分析。
(3)高效:分离效能更进一步。相较于气相色谱和工业精馏塔而言,它的分离效能成倍领先,它可选择流动相或固定相两种不同方式来实现最优的分离结果。
(4)高灵敏度:紫外检测器可达0.01ng,进样量在μL数量级。
(5)应用范围广:对于某些具有强极性、高沸点、大分子、热稳定性差等的店的化合物,他的应用尤为突出。因此它使用于约70%甚至更高比例的化合物。
(6)柱子可反复使用:用一根柱子可分离不同化合物。
(7)样品量少、容易回收:经过色谱柱的反应的样品依旧保持完好,因此可将其回收以做制备或其他用途。
除以上所述之外,样品完好、回收率高、色谱柱重复利用等优点同样给HPLC非常加分。尽管如此,缺点同样不容忽视,如灵敏度相比于气相色谱更低,且有“柱外效应”。所谓的“柱外效应”,即样本在开始检测,但尚未达到检测器时,若流动相的流型发生改变,那么它在诸如柱接头、检测池、进样器、连接管等的柱外空间,被分离物质的些许停滞和分散都会明显地加宽色谱峰,从而降低柱效率。
2.3XRD仪器
2.3.1XRD概念
X射线衍射(简称XRD)是以X射线衍射地方法对物质地图谱进行分析,从而达到明确分子(甚至原子)的组织结构、获取物质组成信息的研究方法。
2.3.2XRD基本理论
作为电磁波的XRD在映射到晶体的过程中会收到类似于从原子中心出发的散射波,这一散射波与源球面波十分相近。原子在晶体中的排列有较强的周期性,这些有固定相位关系的散射球波会加强某方向散射的球面波,以至于形成相互抵消的效果,最终发生衍射现象。就像不同人有不同的指纹一样,每个原子的衍射花样也因其在晶体内部唯一的排列方式而具有不二性(晶胞的位向、大小和形状决定了衍射花样中衍射线的分布规律,而原子的种类及位置决定衍射线的强度),所以可进行物相分析。
参考文献
[1].周志茂.氨噻肟酸结晶过程研究[D].北京化工大学,2011.
[2].于文州,段大成.对我国内酰胺类抗生素发展的建议及十大城市用药情况分析[J].国外医药抗生素分册,2001.22(4):145~150.
[3].Durekheimer W.Recent development of-lactam antibiotics[J].Angewandte Chemic(International Exlition in English),1995,24:180~202.
[4].王荣耕.氨噻肟酸与AE—活性酯[J].精细与专用化学品,2001,(10):18~19.
[5].王荣耕.头孢类抗生素用侧链市场分析[J].精细于专用化学品.2000,(7):11~12.
[6].Rene H,Andre L.3-Acetoxymethyl-7-(hydroxyiminoacetamido)-cephalosporanic acid derivatives.[P].US:4283396,1981-08-11.
[7].肖海焕,黎振球,宋玉叶.氨噻肟酸与AE-活性酯合成的研究进展[J].河南化工,2008(25).
[8].Hong-WooLee,TaeWoe Kang[J].Synthetic Comminucations,1998,28(1):35-44
[9].Kumar,Kyatendra;Arora.et al,Improved preparation of cefpodoxime acid.Indian IN 189315 A 8 Feb 2003,13 pp.(English).(India).
[10].Ochiai Michihiko.2-aminothiazle-4-ylglycine derivative.[P].JP58131975,1983208206
[11].傅德,周鸿娟.2-甲氧亚氨基乙酰乙酸乙酯的合成研究[J].化学世界,1997,38(2):88-90
[12].苏为科,和潮洪.医药中间体制备方法第一册,抗菌药中间体(一)[M].北京,化学工业出版社,2001,358-364.
[13].朱宝泉,李安良,杨兴中等.新编药物合成手册,上卷[M].北京,化学工业出版社,2003,382-383
[14].李从宝,赵美发.氨噻肟酸的生产和应用上[J].化工科技场,2002,10:15-16
[15].朱赛芬,严招春.氨噻肟酸生产技术与应用[J].化工生产与技术,2001,8(5):39-41
[16].张小林,梁兵,全水清等.氨噻肟酸及其衍生物合成新工艺的研究[J].南昌大学学报,2000,22(3):93-96
下载提示:
1、如文档侵犯商业秘密、侵犯著作权、侵犯人身权等,请点击“文章版权申述”(推荐),也可以打举报电话:18735597641(电话支持时间:9:00-18:30)。
2、网站文档一经付费(服务费),不意味着购买了该文档的版权,仅供个人/单位学习、研究之用,不得用于商业用途,未经授权,严禁复制、发行、汇编、翻译或者网络传播等,侵权必究。
3、本站所有内容均由合作方或网友投稿,本站不对文档的完整性、权威性及其观点立场正确性做任何保证或承诺!文档内容仅供研究参考,付费前请自行鉴别。如您付费,意味着您自己接受本站规则且自行承担风险,本站不退款、不进行额外附加服务。
原创文章,作者:写文章小能手,如若转载,请注明出处:https://www.447766.cn/chachong/14702.html,